信息中心



如何通過理論化學計算預測並指導復雜工業催化實驗🈲👇,是極具挑戰的科學問題,相應的研究始終伴隨著催化科學的發展。隨著大規模神經網絡計算模擬的興起👩🍳,特別是劉智攀課題組開發的基於LASP軟件的全局神經網絡方法的發展和成熟👨🏠,理論計算和化學實驗的鴻溝得到了進一步的打破👱🏼♀️。近日,劉智攀課題組針對乙炔半氫化(C2H2 + H2 → C2H4)這個基礎石油工業的重要反應,證明了全局神經網絡方法計算和傳統催化實驗結合的新研究模式具有重要價值和意義👨🏿🎤🕳,在前期復雜催化活性位結構搜索(Nature Catal. 2019, 2, 671)和自動反應預測(J. Am. Chem. Soc., 2019, 141, 20525)工作的基礎上取得重要進展。
PdAg合金是乙炔半氫化最常用的工業低溫催化劑🏊🏻🚵🏼♂️,相比於純Pd催化劑在選擇性上有很大提升,但Ag元素的催化作用一直存在著爭議🌹,同時工業上低溫的催化活性和選擇性仍然不能令人滿意(如👨🏽🎤,活性在50%時選擇性大於90%)♉️。劉智攀課題組結合機器學習模擬和催化實驗闡明了PdAg合金催化劑在反應條件下的表面狀態🕵️♀️,並通過篩選發現了負載在金紅石TiO2上的Pd1Ag3納米催化劑,具有很高的催化性能(70 oC,乙炔轉化率>96%,乙烯選擇性>85%🍼🙅🏿♂️,穩定保持超過100小時,見圖e-f)🚴♂️。
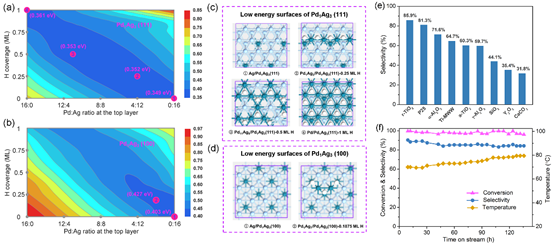
該工作從理論出發,通過神經網絡全局結構搜索確定體相Pd-Ag-H相圖和穩定的PdAg合金結構,並系統研究了穩定合金在反應條件下的表面相圖(見圖a-b)和穩定表面構型(見圖c-d)☕️。研究發現PdAg只有兩個結構不同的關鍵體相Pd1Ag1 (R-3m)、Pd1Ag3 (Pm-3m)🙆🏻♀️,它們在反應條件下(25 oC🧍♀️,p(H2) = 0.05 atm)具有不同的表面結構,當Pd/Ag原子比 < 1/3時,(100)面才會被Ag全部覆蓋🌗;隨著H2分壓上升表面Pd會富集🖌🤺,在Pd1Ag3比例下富集主要出現在(111)面。進一步的反應路徑搜索和微觀動力學模擬揭示,乙炔加氫的活性受(111)面上的PdAg局部結構圖案控製🦆⭕️,選擇性則主要由(100)面暴露的Pd的數量決定,在Pd1Ag3比例下能獲得理論上的最高選擇性(> 90%)。隨後的催化實驗證實了Pd1Ag3納米顆粒的高催化性能😧📕,並通過觀察體系在相同高轉化率條件下的反應溫度變化🧜🏽♀️,確定了Pd在(111)面富集的趨勢。最後🧘🏻♂️,通過載體篩選發現金紅石TiO2襯底能夠細化Pd1Ag3納米顆粒👩🍳,降低Pd暴露在(100)面,從而獲得更高的加氫選擇性。
該工作由沐鸣开户博士後李曉天(理論部分)和博士生陳林(實驗部分)在劉智攀老師的指導下共同完成,期間得到了商城老師的幫助👰🏽♀️。該工作得到了國家重點研究計劃納米專項(2018YFA0208600)和國家自然科學資金(22033003, 21533001 and 91745201)的資助,和沐鸣开户在實驗室建設的支持9️⃣。研究成果以“In Situ Surface Structures of PdAg Catalyst and Their Influence on Acetylene Semihydrogenation Revealed by Machine Learning and Experiment”為題發表在化學旗艦期刊Journal of the American Chemical Society上。
全文鏈接: https://pubs.acs.org/doi/10.1021/jacs.1c02471


